
Nephrology: Translational
Category: Abstract Submission
Nephrology I: Potpourri
 photo")
Laurel Willig, MD, MS (she/her/hers)
Pediatric Nephrology, Associate Professor
Children’s Mercy Kansas City
Kansas City, Missouri, United States
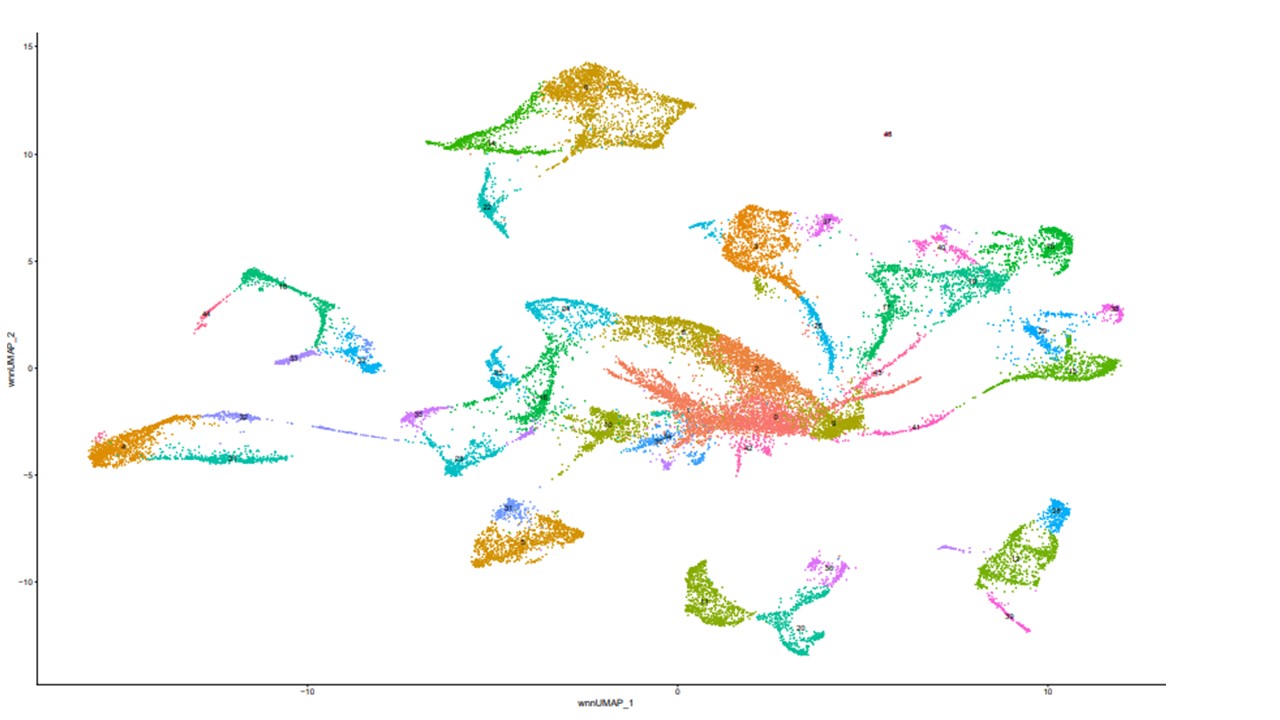 17 clusters were identified using marker genes as belonging to the renal tubular compartment. These clusters include the following: 2, 3, 8, 9, 13-19, 24-26, 29, 37, 38, and 43.
17 clusters were identified using marker genes as belonging to the renal tubular compartment. These clusters include the following: 2, 3, 8, 9, 13-19, 24-26, 29, 37, 38, and 43..jpg) Gene ontology terms associated with genes that are upregulated in PKD1 +/- tissue compared to either wildtype tissue, minimally affected tissue from PKD1 +/- pigs, or in genes upregulated in both comparisons (ie genes upregulated in the PKD1+/- tissue in both analyses).
Gene ontology terms associated with genes that are upregulated in PKD1 +/- tissue compared to either wildtype tissue, minimally affected tissue from PKD1 +/- pigs, or in genes upregulated in both comparisons (ie genes upregulated in the PKD1+/- tissue in both analyses).