Icon Legend
This session is not in your schedule.
This session is in your schedule. Click again to remove it.
Presentation Icons

Pre-Conference Ticketed Event

AAP

APA George Armstrong

APA Health Care Delivery Award

APA Global Health Research Award

Miller Sarkin Mentoring Award

David G. Nathan Award (Fellow Basic)

Fellow Basic Research Awards

Fellows Junior Section Basic Research Award

Fellows Junior Section Clinical Research Award

Fellow Clinical Research Awards

House Officer Research Awards

Japan Pediatric Society Fellow Exchange Awards

Maureen Andrew Mentor Award

New Member Outstanding Science Award

SPR Bridging to Success Award

SPR Diversity in the Research Workforce Travel Award - ESPR

Richard D. Rowe Award (Fellow Clinical)

SPR Award in honor of E. Mead Johnson

Student Research Awards

Young Investigator Award

Norman J. Siegel New Member Outstanding Science Award

John Howland Award

Trainee Research Award-Clinical

Trainee Research Award-Basic

Richard D. Rowe Award

Mary Ellen Avery Award

PAS 2022 Lab

SPR Thomas A. Hazinski Distinguished Service Award

SPR Douglas K. Richardson Award for Perinatal and Pediatric Healthcare Research

PROSPER Diversity Award


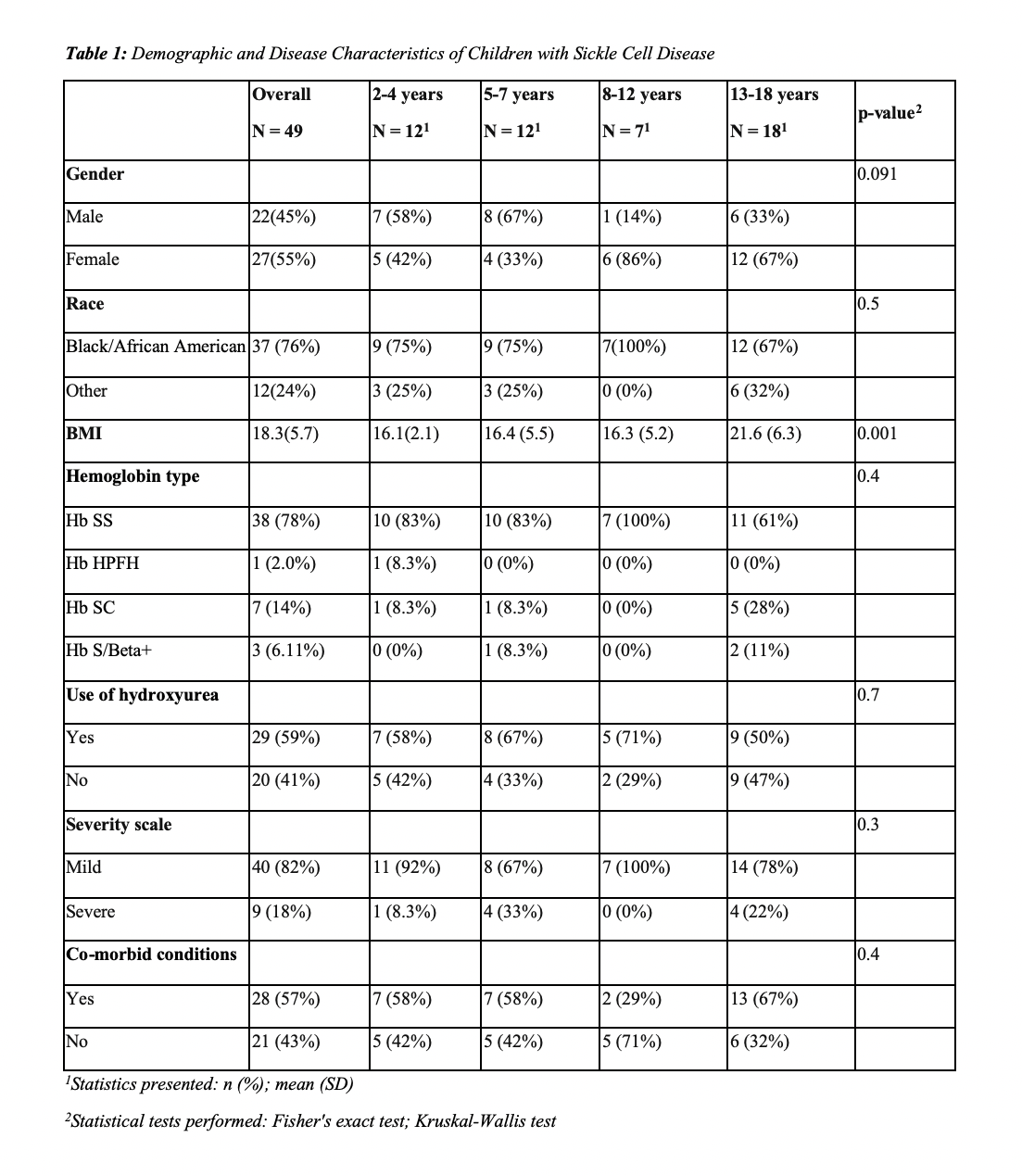 1Statistics presented: n (%); mean (SD)
1Statistics presented: n (%); mean (SD)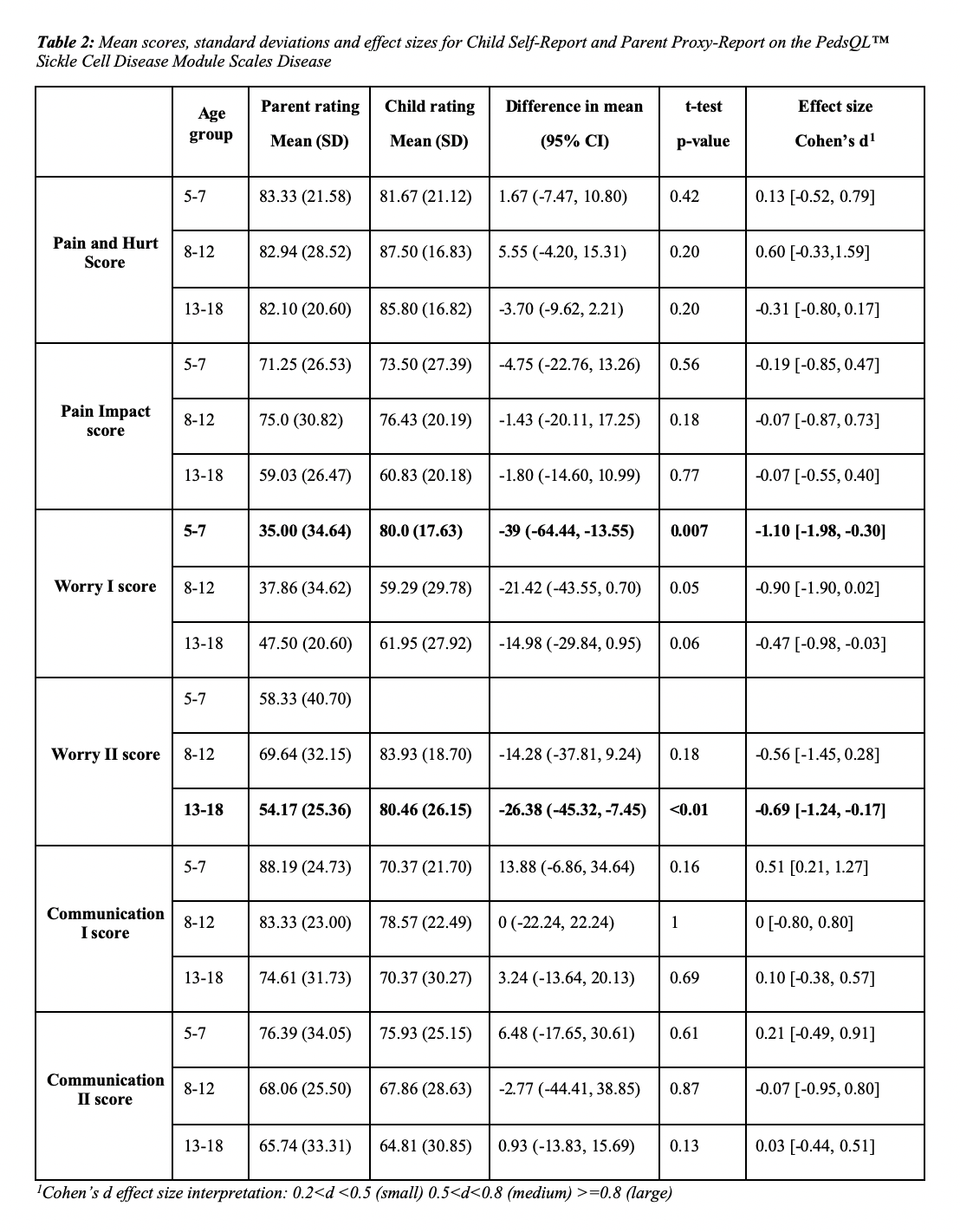 1Cohen’s d effect size interpretation: 0.2=0.8 (large)
1Cohen’s d effect size interpretation: 0.2=0.8 (large)