
Genomics/Epigenomics
Category: Abstract Submission
2: Genomics/Epigenomics II

Jim N. Jarvis, MD
Professor of Pediatrics
Jacobs School of Medicine and Biomedical Sciences at the University at Buffalo
Buffalo, New York, United States
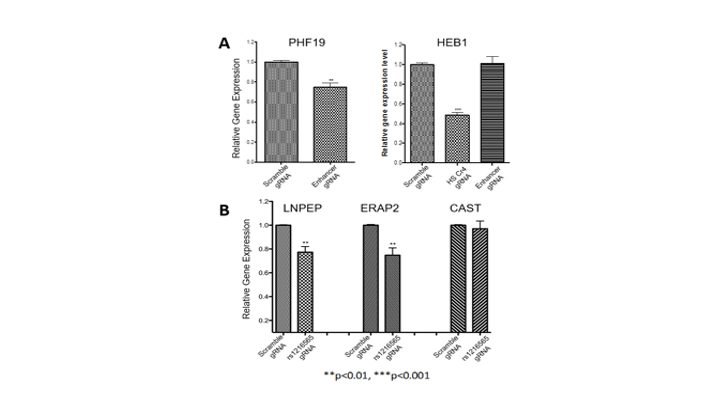 For each experiment, 4 gRNAs were targeted to the functional regions of intergenic enhancers in the TRAF1 locus and the ERAP2/LNPEP locus. Bar graphs summarize the results of 4 independent experiments. (A) Attenuation of the intergenic enhancer at the TRAF1 locus significantly reduced expression of PHF1, but not C5 (not shown). Off-target effects are monitored by showing that these gRNAs have no effect on HEB1 expression, but that gRNAs directed to an enhancer known to regulated HEB1 attenuates expression. (B) Attenuating the intergenic enhancer in the LNPEP/ERAP1 locus reduces expression of both LNPEP and ERAP2, but not the adjacent gene, CAST, nor the HEB1 gene (not shown). In each experiment, scrambled versions of the gRNAs had no effect on expression of the putative targets.
For each experiment, 4 gRNAs were targeted to the functional regions of intergenic enhancers in the TRAF1 locus and the ERAP2/LNPEP locus. Bar graphs summarize the results of 4 independent experiments. (A) Attenuation of the intergenic enhancer at the TRAF1 locus significantly reduced expression of PHF1, but not C5 (not shown). Off-target effects are monitored by showing that these gRNAs have no effect on HEB1 expression, but that gRNAs directed to an enhancer known to regulated HEB1 attenuates expression. (B) Attenuating the intergenic enhancer in the LNPEP/ERAP1 locus reduces expression of both LNPEP and ERAP2, but not the adjacent gene, CAST, nor the HEB1 gene (not shown). In each experiment, scrambled versions of the gRNAs had no effect on expression of the putative targets.